Cystisk fibrose er en medfødt metabolisk sygdom. Kropsvæsker såsom spyt, bronkial slim eller bugspytkirteludskillelse er meget hårdere end normalt på grund af genetisk disponering. Konsekvenser inkluderer åndedrætsproblemer og fordøjelsesbesvær. Cystisk fibrose kan ikke hærdes. Med konsistent terapi kan sygdomsforløbet imidlertid nedsættes. Læs her, hvilke symptomer der forårsager cystisk fibrose, og hvordan du behandler den.

Cystisk fibrose: kort oversigt
- Beskrivelse: arvelig metabolisk sygdom, der forårsager hård slimdannelse i lungerne og andre organer
- symptomer: Luftvejsproblemer, irriterende hoste, lungeinfektion, manglende trivsel, fordøjelsesbesvær, svær diarré, fedtlever, nedsat fertilitet
- årsager: Arv af defekte gener, der påvirker konsistensen af kropsvæsker, udbrud af sygdommen kun, hvis begge forældre arver et sygt gen (dominerende recessiv arv)
- diagnose: Blodprøve for immunreaktivt trypsin (IRT), pancreatitis-associeret protein (PAP), svedtest, genetisk test
- behandling: mukolytiske midler, bronchodilaterende midler, inhalation, antibiotika til infektioner, kortison, CFTR-modulatorer, lungetransplantation
- prognose: ikke helbredelig, naturligvis stærkt afhængig af alvorligheden og tidspunktet for diagnosen, forkortet forventet levealder
Cystisk fibrose: beskrivelse
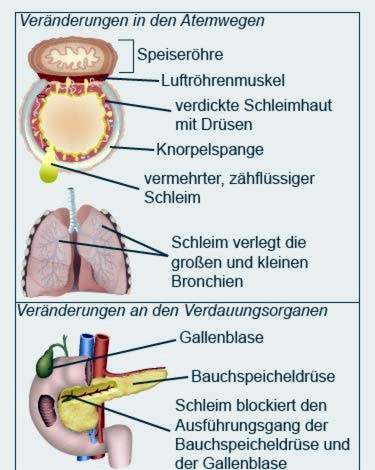
Cystisk fibrose (også kaldet cystisk fibrose) er en arvelig metabolisk sygdom. Dannelsen af forskellige kropsvæsker forstyrres. Sekretionerne i lungerne, bugspytkirtlen og andre organer er mere tyktflydende end hos raske mennesker.
Hårdt slim
Det hårde slim tilstopper blandt andet de små grene i bronchierne og kanalerne i de indre organer. Åndedræt og fordøjelse påvirkes især. I løbet af sygdommen kan organerne fungere værre og værre.
Fejl i det genetiske materiale
Årsagen til sygdommen er defekter i det genetiske materiale. Cystisk fibrose kan derfor ikke hærdes. Diagnostikstid og symptomens sværhedsgrad kan variere meget individuelt. Hos mange børn har cystisk fibrose en massiv indflydelse fra fødslen, i andre tilfælde anerkendes det senere.
Cystisk fibrose: symptomer
Cystisk fibrosesymptomer kan variere meget fra patient til patient. Sygdommen påvirker funktionen af forskellige organer, men især lungerne og fordøjelsessystemet.
Genkend tidlige tegn
De første symptomer på cystisk fibrose varierer individuelt. I de fleste tilfælde vises cystisk fibrosesymptomer inden for det første leveår. Sygdommen kan således normalt diagnosticeres tidligt og startes hurtigt med en terapi. Nogle patienter har imidlertid kun betydelige klager i ungdomsårene. Ikke alle berørte personer viser hele spektret af mulige symptomer. Alvorligheden af symptomerne varierer også.
Ændrede kropsvæsker
Ved cystisk fibrose forstyrres dannelsen af de såkaldte chloridionkanaler i cellerne. Dette ændrer sammensætningen af kropsvæsker. Den nemmeste måde at opdage denne ændring i sved hos de berørte. Din sved er saltere end raske mennesker. Saltene natrium og chlorid, der hører til de såkaldte elektrolytter, er beriget med deres sved. Som et resultat af svedtab mister patienter, der lider af cystisk fibrose, i stigende grad kropssalte.
Varierede symptomer på cystisk fibrose
Sygdommen påvirker en lang række organsystemer. Ofte vises de første symptomer på cystisk fibrose i lungerne og fordøjelseskanalen. I løbet af livet kan der tilføjes yderligere klager. Gennem målrettet terapi kan symptomerne behandles godt. Imidlertid kan symptomerne også nå en truende proportioner. Det er især farligt, når bronchierne tilstoppes gennem det viskose slim. Derefter kan patienterne kvæle i ekstreme tilfælde.
Cystisk fibrose: symptomer på lungen
Åndedrætsproblemer og irriterende hoste
I de fleste tilfælde forekommer symptomer på lungerne kun i lidt ældre spædbørn ved cystisk fibrose. Nyfødte har normalt ingen åndedrætsproblemer. Cystisk fibrosesymptomer udtrykkes ofte i form af en hoste-halslignende, kronisk, irriterende hoste hos lidt ældre børn. Slimet i deres luftvej er øget, hårdt og tyktflydende. Dette hindrer luftstrømmen i lungerne. Over tid udvikler sig en progressiv åndedrætsbesvær.
Hyppige infektioner
Øget slimproduktion i lungerne gør det lettere for bakterier at kolonisere og forårsage infektion. Gentagende lungebetændelse eller bronchiale infektioner er hovedsageligt forårsaget af bakterier såsom stafylokokker og Pseudomonas-arter. Den forstyrrede saltbalance i lungerne hindrer også kroppens forsvar. Der kan også forekomme lungeblødninger. Et typisk tegn på dette er hoste af blodblandet slim.
Selvom lungerne er beskadiget fra en tidlig alder, er de første symptomer på cystisk fibrose i luftvejene ofte kun i grundskolealderen eller endda senere. Symptomerne opstår undertiden kun, når store dele af lungevævet allerede er blevet ødelagt, eller luftvejene er hårdt indsnævret.
Cystisk fibrose: symptomer på bugspytkirtlen
Hos patienter med cystisk fibrose bliver bugspytkirtlen ofte betændt. Den udskiller en sekretion, der blandt andet indeholder enzymer til fordøjelse af fedt og sukker. Hos patienter med cystisk fibrose kondenseres sekretionen på grund af dens viskositet tilbage og forårsager betændelse.
Når processen skrider frem, bliver pancreasvævet hærdet og arret. Læger taler om fibrose. Fibrosen ødelægger gradvis bugspytkirtlen. Ud over galden danner bugspytkirtlen også insulin, som er nødvendigt blandt andet til sukkerudnyttelsen i kroppen. Patienter, der er lidt ældre (fra ungdomsårene), udvikler ofte diabetes mellitus.
Cystisk fibrose: symptomer på galden
Bugspytkirtlen og galdeblæren deler en fælles kanal i tarmen. Derfor kan tilbagestrømningen af pancreasudskillelser også forårsage betændelse i galdeblæren. Ofte dannes gallesten, som fuldstændigt kan blokere galdeblæren fra galdeblæren.
Cystisk fibrose: symptomer på fordøjelseskanalen
Ud over klager over lungerne påvirker cystisk fibrosesymptomer hovedsageligt fordøjelsen. På grund af den manglende galde, for eksempel, forringes fedtfordøjelsen. Ofte tolererer patienter fedtholdige fødevarer dårligt. Den indtagne mad udskilles stort set ufordøjet igen. Typiske er derefter meget omfangsrige og bløde tarmbevægelser.
Diarré og vækstlidelser
Berørte børn, inklusive spædbørn, lider ofte af svær diarré. Selvom de drikker og spiser godt, øges de næppe. Vækstlidelser og underernæring er derfor yderligere klassiske konsekvenser af sygdommen.
Imidlertid kan sådanne klager inden for fordøjelse også forekomme i andre sygdomme. Kun i kombination med åndedrætsproblemer er de derfor en karakteristisk indikation af cystisk fibrose, som bestemt bør undersøges.
anal prolaps
I det videre forløb kan cystisk fibrose forårsage forskellige komplikationer i fordøjelseskanalen. Den mest almindelige er en såkaldt anal prolaps. Ved anal prolaps bukker tarmslimhinden ud af anus. En sådan hændelse skal behandles kirurgisk så hurtigt som muligt.
tarmobstruktion
Involvering af tarmen (invagination) eller intestinal obstruktion (ileus) er også almindelig. Begge komplikationer er forbundet med svær mavesmerter og betydelige fordøjelsesproblemer. Smerten forekommer normalt i spurts, især efter at have spist. En tarmobstruktion er dødelig, hvis den ikke behandles. Krampagtig, akut mavesmerter bør derfor altid afklares af en læge.
Cystisk fibrose: symptomer på leveren
fedtlever
Under tilbagestrømmen af galde lider også leveren. Hos mange patienter udvikles fedtlever i løbet af sygdommen. Træthed, appetitløshed, oppustethed og flatulens og i sjældne tilfælde kan følelser af tryk eller smerter i øvre del af maven forekomme.
skrumpelever
I sjældne tilfælde udvikler sig en krympende lever (levercirrhose), hvor leveren er alvorligt forstyrret i sin funktion. Det manifesterer sig først i form af gulsot (gulsot). Tegn på gulsot er den gulaktige misfarvning af hvidt i øjnene. Efter lang tid forekommer også hjerteproblemer, og ydeevnen for de berørte fortsætter med at falde.
Cystisk fibrose: reduceret fertilitet
Mere end halvdelen af alle mandlige patienter er infertile. Selv om de i de fleste tilfælde kan danne frugtbar sæd, kan de ikke komme igennem vas deferensne, fordi de er blokeret af tyktflydende slim.
Berørte kvinder er normalt mindre frugtbare. Generelt kan de modtage og levere et barn. Imidlertid akkumuleres hårdt slim i deres æggeledere, som sædcellen næppe kan trænge ind i. Især i en højere alder falder sandsynligheden for graviditet hurtigt.
Cystisk fibrose: symptomer hos børn
Cystisk fibrose er en genetisk sygdom. Det har altid været til stede siden fødslen. Men de klassiske symptomer på cystisk fibrose forekommer ikke altid i barndommen. Der er dog ofte allerede uspecifikke instruktioner, der skal følges. Dette gælder især, hvis der allerede er opstået tilfælde af cystisk fibrose i familien.
Oppustet mave
En indikation af, at der er en metabolisk lidelse, er for eksempel en oppustet mave i lang tid. Ofte lider børnene af diarré. Hos nyfødte kan en markant forsinket første afføring (Kindspech) være en indikation af cystisk fibrose. I mange tilfælde forekommer vækst- og vækstlidelser, selvom børnene spiser med trang. Kun i sjældne tilfælde fører det også til forstoppelse (forstoppelse) som et resultat af sygdommen.
Rattling vejrtrækning
Andre symptomer, der kan indikere cystisk fibrose, inkluderer skramlende vejrtrækning og alvorlig rastløshed. Mange børn lider af kronisk betændelse i bihulerne. Disse mærkes hovedsageligt ved ikke nøjagtigt lokaliserbar smerte i ansigtet. Nasale polypper er mere almindelige hos børn med cystisk fibrose end hos raske børn.
Hvis børn lider af åndedrætsproblemer eller fordøjelsesbesvær, skal en læge altid konsulteres som en forsigtighedsforanstaltning. Hos børn kan livstruende situationer hurtigt opstå, fordi de ikke selv kan formulere deres klager, eller deres sværhedsgrad ikke kan vurdere.
Cystisk fibrose: årsager og risikofaktorer
Cystisk fibrose er forårsaget af en genetisk defekt. Den patologiske ændring ligger på det syvende kromosom i det såkaldte CFTR-gen.
CFTR-genet (cystisk fibrose-transmembranregulatorgen) indeholder konstruktionsmanualen til en kanal, gennem hvilken chloridioner kommer ind i cellerne. De mangelfulde chloridionkanaler blokerer transporten af salt ind i visse kropsceller hos cystisk fibrosepatienter.
De berørte kirtelceller, men i stedet for det ellers flydende sekretion, er hårdt slim fra. I lungerne danner paranasale bihuler, bugspytkirtlen, tarmen, galdekanalerne og gonaderne så hårde slimudskillelser med høj saltholdighed.
Cystisk fibrose: hvor truet er mit barn?
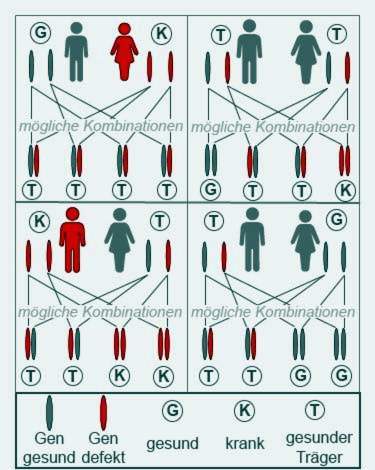
Cystisk fibrose bryder kun ud, når begge forældre videregiver et patologisk ændret gen til deres barn. Disse forældre er da for det meste begge selv sunde, men bærere af genet.
Mennesker med cystisk fibrose er kun delvist frugtbare. Nogle patienter bliver stadig forældre. Syge fædre eller mødre videregiver imidlertid altid et sygt gen, da begge CFTR-gener bærer cystisk fibroseinformation. Deres børn bliver dog kun syge, hvis de også får et sygt gen fra den anden forælder.
Par, hvis familier allerede har haft tilfælde af cystisk fibrose, skal søge genetisk rådgivning inden de planlægger en graviditet.
Ægsortering
Forældre, der kunne føde cystisk fibrose, kan gennemgå genetisk diagnose af præimplantation. Ved genetisk testning af preimplantation befrugtes oocytterne først kunstigt. De første celledelinger finder sted i prøverøret (in vitro).
Før der indsættes et embryo, testes det for ændrede genegenskaber. Derefter implanteres der kun embryoner, der ikke bærer det syge gen. Uanset dette kan det også undersøges under graviditet, om barnet senere vil udvikle CF.
Cystisk fibrose: undersøgelser og diagnose
I modsætning til for et par år siden gennemgår de fleste hospitaler i dag rutinemæssigt neonatal screening. Dette inkluderer undersøgelser af cystisk fibrose.
Screening i blod og sved
Til screening tages blod fra den nyfødte. Cystisk fibrostest involverer flere trin:
blodprøve
Test for forhøjede niveauer af immunreaktivt trypsin (IRT) og pancreatitis-associeret protein (PAP). I tilfælde af abnormiteter finder svedtesten sted.
svejsning test
Cystisk fibrosepatienter har en signifikant højere saltholdighed hos sved end raske mennesker. Ved cystisk fibrose-svedtest måles indholdet af salte natrium og klorid i kropssved. Hos børn opsamles sveden på underarmen og analyseres derefter. Hvis der opstår mistanke herom, udføres en genetisk test.
Genetisk test
Hos patienter med cystisk fibrose er det såkaldte CFTR-gen, der giver planen for specifikke ionkanaler, ændret. Denne konstruktionsmanual er lang, den består af ca. 6500 basepar. Overalt kan en fejl krybe ind i kode, men fejlene har forskellige effekter. Derfor testes kun de mest almindelige afvigelser i koden.
Familiehistorie giver tip
Hvis der ikke er udført nogen passende neonatal screening, og den mistanke om cystisk fibrose dukker op senere, er familielægen eller internisten den rigtige person til at kontakte. I en indledende samtale registrerer denne person den medicinske historie (anamnese). I tilfælde af mistanke om cystisk fibrose er der særlig opmærksomhed på familiehistorien.
Fysisk undersøgelse
Derefter finder en fysisk undersøgelse sted. Lægen lytter til lungerne og scanner de indre organer. Han kan allerede udelukke nogle andre tilstande, der er forbundet med symptomer, der ligner symptomer på cystisk fibrose.
Derudover kan røntgenundersøgelser vise respirations- og lungeblokkinger. Laboratorieundersøgelser giver indikationer af funktionelle begrænsninger af de indre organer. Selv hos voksne, der er mistænkt for cystisk fibrose, giver en svedtest vigtige beviser for diagnosen.
Testning af familiemedlemmer
Hvis der påvises cystisk fibrose i en familie, giver det mening at alle andre familiemedlemmer også gennemgår en undersøgelse. Cystisk fibrose kan også forekomme i svækkede former. Det tager ofte mange flere år for cystisk fibrose at manifestere sig med klare symptomer. Ikke desto mindre er tidlig diagnose og cystisk fibroseterapi også vigtig for dem, der er berørt for at øge forventet levealder.
Cystisk fibrose: behandling
Cystisk fibrose kan ikke hærdes. Børn født med cystisk fibrose lider af sygdommens virkninger i hele deres liv. Imidlertid kan en kombination af fysioterapi, medicin og indånding betydeligt nedsætte sygdommens progression. En cystisk fibroseterapi bør derfor primært opnå, at de berørte kan føre et så normalt liv som muligt.
At lære at leve med sygdommen
Især for børn er det vigtigt at lære at håndtere sygdommen på lang sigt. Børn med cystisk fibrose bør lære så tidligt som muligt, hvad sygdommen betyder, og hvordan den påvirker kroppen. Her kan et hospitalophold med specielle træningsenheder være nyttigt. I processen lærer børn og forældre, hvordan de skal fodre sig selv, hvordan sport ser ud og hvordan de opfører sig bedst i kritiske situationer.
Hjælp til lungerne
Der er forskellige muligheder for behandling af symptomerne. Afhængig af patientens alder og sværhedsgraden af symptomerne anbefales forskellige tilgange.
Mukolytiske midler
Cystisk fibrosepatienter lider mest af lungeproblemer. Ved regelmæssig indånding med specielle tilsætningsstoffer (mucolytika) opløses det viskose slim og kan let hostes af.
Bronchiale dilateringsmidler
Såkaldte beta-2-sympatomimetika øger også bronchierne, hvilket yderligere letter vejrtrækningen.
Antibiotika mod bakterier
På grund af den dårlige ventilation i lungerne lider mennesker med cystisk fibrose af bakterieinfektioner i luftvejene meget hyppigere. På den anden side hjælper antibiotika, der indgives i god tid. I nogle tilfælde giver det mening at indånde disse permanent.
Antiinflammatoriske lægemidler
Hos mange patienter er luftvejene ofte eller kronisk betændte. Derefter hjælper antiinflammatoriske lægemidler som kortison.
CFTR modulatorer
I mellemtiden er de første lægemidler blevet udviklet, der forbedrer ionkanalernes nedsatte funktion. De arbejder imidlertid kun for visse mutationer og dermed kun for en lille del af patienterne. Deres effektivitet er også begrænset. Forbedring af lægemidlerne undersøges intensivt. For første gang begyndte de med årsagen til sygdommen snarere end blot med symptomerne.
Lungetransplantation – det sidste håb
I alvorlige tilfælde er en lungetransplantation også en mulighed. Så drastisk som dette trin ser ud til at begynde med, kan mange patienter leve et markant lavere belastet liv bagefter.
Fød korrekt i cystisk fibrose
Da cystisk fibrose også forstyrrer fordøjelsen, skal patienterne være nøje opmærksomme på deres kost. Du skal foretrække en diæt med protein og kulhydrater.
Der er også vitamintilskud og mineraler. Sidstnævnte erstatter salte, der sveder patienterne i store mængder. Da bugspytkirtlen ikke fungerer korrekt, får børn medicin til fordøjelsesenzymer som et supplement til måltider.
Oplev vaccinationer
Vaccination er især vigtig for patienter med CF. Hos dem har bakterier lettere leg, og de bliver ofte mere alvorlige end patienter, der ikke er forhåndsindlæst. Specielt vacciner mod mæslinger og pneumokokker anbefales. Derudover bør hvert år være en influenzavaccine.
Cystisk fibrose: sygdomsforløb og prognose
Cystisk fibrose er forårsaget af en ændring i genomet og kan derfor ikke hærdes. For mennesker med cystisk fibrose reduceres forventet levetid og livskvalitet normalt markant. Uden terapi forværres sundhedstilstanden hurtigt, og de berørte lever normalt ikke længe.
Med rettidig og konsistent terapi kan sygdommens forløb nedsættes betydeligt. I mellemtiden lever patienter meget længere end for et par år siden. Den gennemsnitlige forventede levealder for cystisk fibrose er i øjeblikket omkring 40 år. Men mange lever også med sygdommen i 50 år og mere.
Komplikationer og følger
Selv ved intensiv terapi kan komplikationer forekomme igen og igen ved cystisk fibrose. Oftest er der akut åndedrætsbesvær på grund af dårlig lungeventilation. Individuelle områder i lungen kan endda kollapse (alektase).
Ofte udvikles kronisk bronkitis eller lungebetændelse. Svampe kan også påvirke lungerne.
Derudover kan forskydninger i væske- og elektrolytbalancen udløse chok og kredsløbssvigt.
Andre komplikationer og følger er:
- kronisk leversygdom, især cirrose
- Galdeblærebetændelser og galdesten
- kronisk betændelse i bugspytkirtlen
- forstyrret hjertefunktion
- akut tarmobstruktion (ileus)
- Intestinal invagination
- underernæring
- Diabetes mellitus
- Begrænset fertilitet hos kvinder eller infertilitet hos mandlige patienter
Cystisk fibrose er en arvelig sygdom, forebyggelse er derfor ikke mulig. Mennesker med risiko for familiær risiko skal søge genetisk rådgivning, hvis de ønsker at få børn. En genetisk test udføres, og det undersøges, om CFTR-genet er ændret. Afhængig af om en eller begge partnere bærer genet, kan risikoen for afkommet beregnes.
I mellemtiden er genetisk diagnose (PID) i cystisk fibrose også mulig i Tyskland. Forudsætningen herfor er altid godkendelse af et etisk udvalg. Oocytter befrugtes uden for livmoderen og kun embryoner uden de problematiske gener af cystisk fibrose anvendes.
Yderligere information
retningslinjer:
- S2 konsensusretningslinje “Diagnostik af cystisk fibrose” fra Society for Pediatric Pulmonology (2013)
- S3-retningslinje “Lungesygdom i cystisk fibrose” af Society for Pediatric Pulmonology (2013)
Support Grupper:
- Cystisk fibrose e.V.